GROMACS
Created by [ Potthoff, Sebastian], last modified on 23. Jun 2023
Gromacs is one of the most commonly used molecular dynamics codes running on almost every HPC cluster. However there are a few things to know about running Gromacs efficiently.
Use the \"gmx_mpi\" binary
The Gromacs modules on PALMA come with two installed binaries:
- gmx
- gmx_mpi
Use the gmx_mpi binary. The gmx binary is the threadedMPI version of Gromacs and is not suitable for multi-node jobs.
Stick with MPI parallelism
Even though Gromacs supports OpenMP parallelism, it is easiest to stick to pure MPI parallelism. This can be achieved by setting the number of OpenMP threads to one per MPI task either via the environment variable OMP_NUM_THREADS or via the Gromacs flag --ntomp.
Do not use simultanious multi-threading aka. \"hyper-threading\"
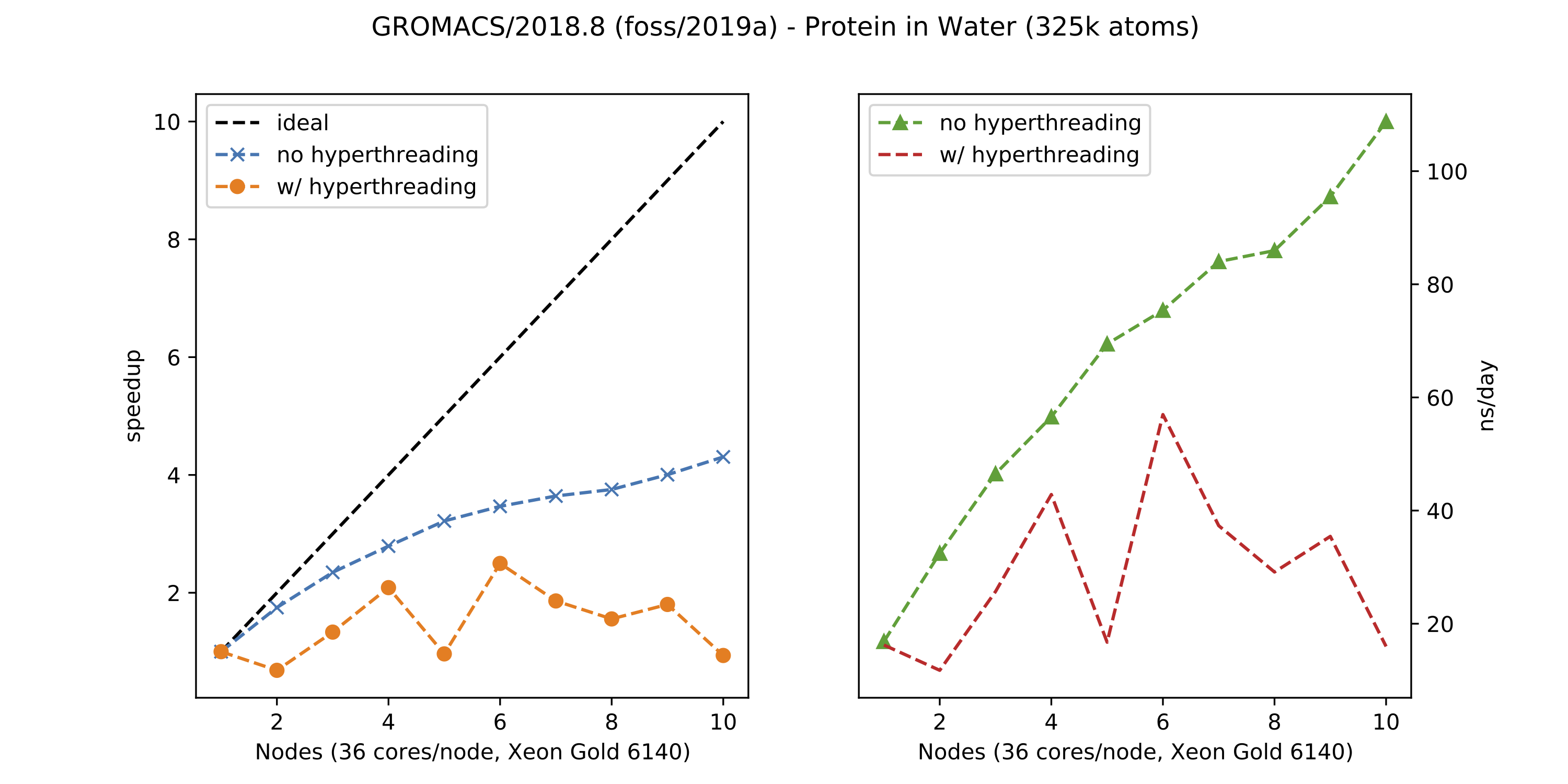
~~Gromacs does not benefit from using all available hardware threads on a node; in fact performance suffers substantially. In the plots below you can see the difference in speedup (and ns/day) between a system with enabled and disabled hyper-threading:~~
[ {.confluence-embedded-image
height="400"}]{.confluence-embedded-file-wrapper
.confluence-embedded-manual-size}
{.confluence-embedded-image
height="400"}]{.confluence-embedded-file-wrapper
.confluence-embedded-manual-size}
~~This means when you request resources, you should always request double the number of tasks (cpus) you actually intend to use and pin Gromac's processes to the cores. Slurm (srun) does this by default but you could also specify --cpu-bind=cores.~~
note: By now we disabled hyper-threading on PALMA. So you can just use the amount of cores you requested.
Example Job Script {#GROMACS-ExampleJobScript}
An example job script acknowledging all the above could look like this
#!/bin/bash
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=36
...
# Load modules
ml palma/2019a
ml foss/2019a
ml GROMACS/2018.8
# Set number of OpenMP threads to 1
export OMP_NUM_THREADS=1
# Start GROMACS
# Note that even though we do not explicitly tell srun how many tasks per node we want to use,
# it automatically distributes them evenly accross all nodes (in this example 36 per node)
srun gmx_mpi mdrun ...
![]() speedup_1-10nodes.pdf
(application/pdf)\
speedup_1-10nodes.pdf
(application/pdf)\
![]() speedup_1-10nodes.png
speedup_1-10nodes.png
{kind=link}